










-
Head & Neck Tumour Surgery

-
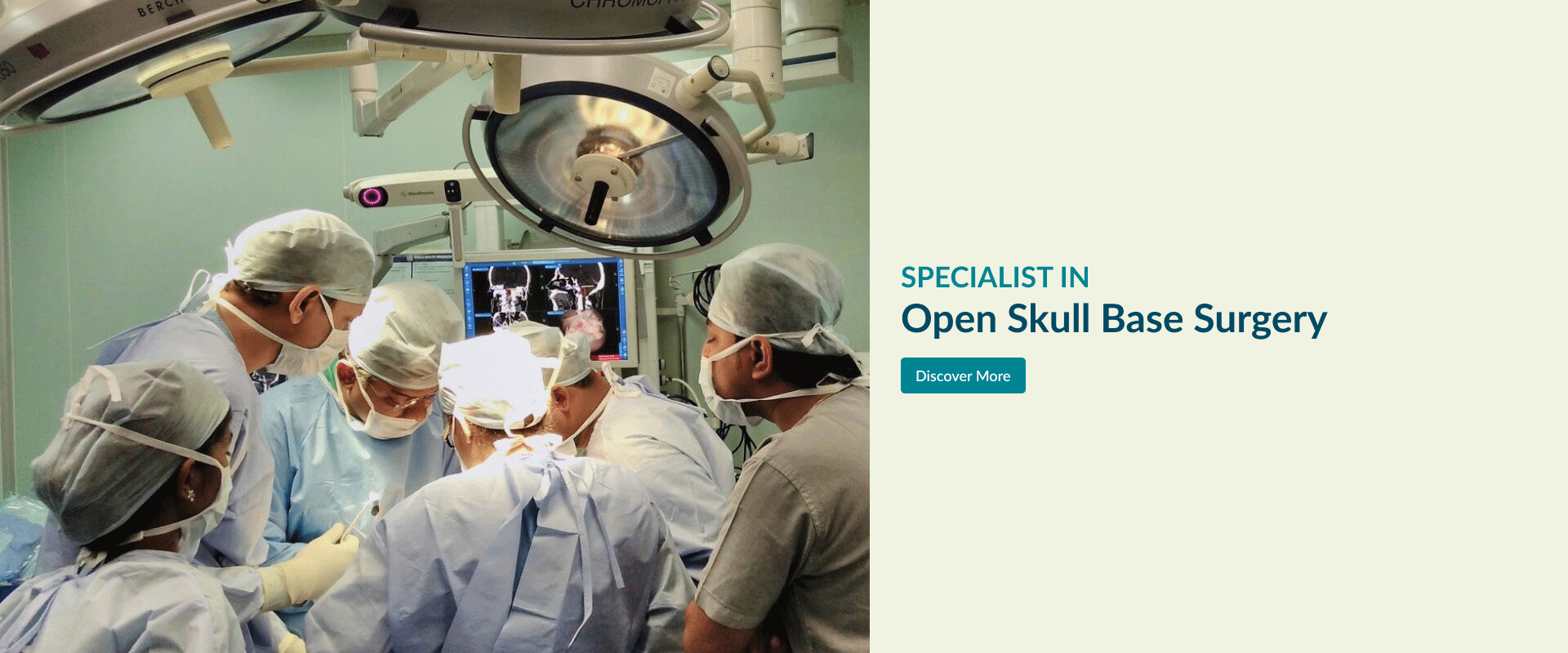
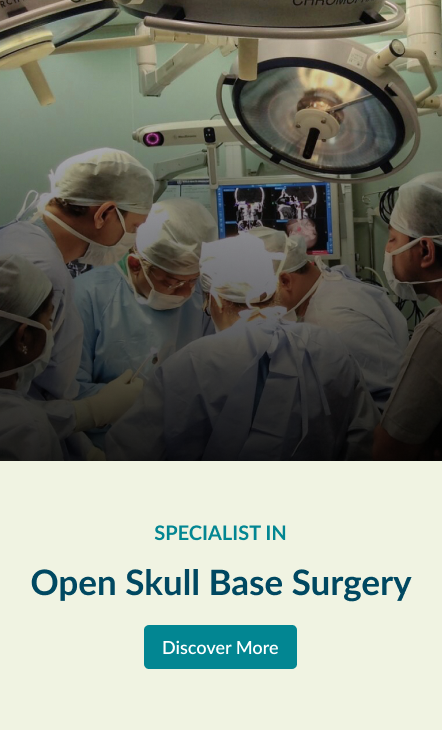
Open Skull Base Surgery
-
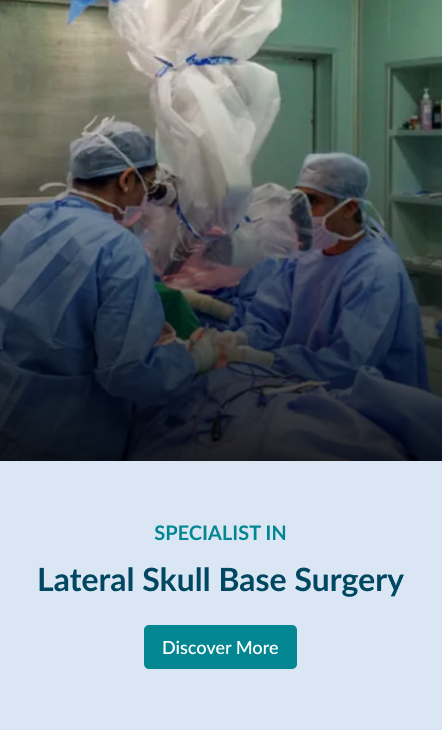
Lateral Skull Base Surgery
-
Endoscopic Pituitary Surgery
-
Endoscopic Skull Base Surgery
-
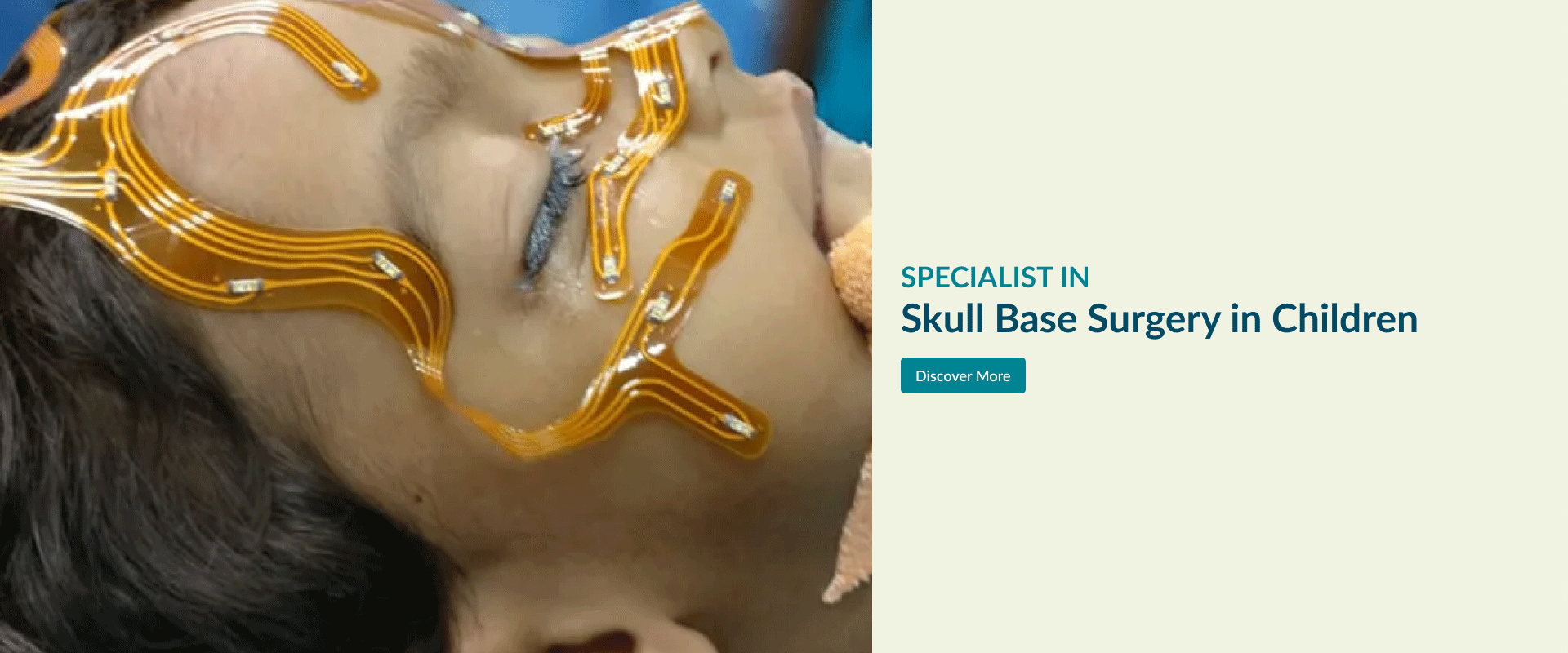
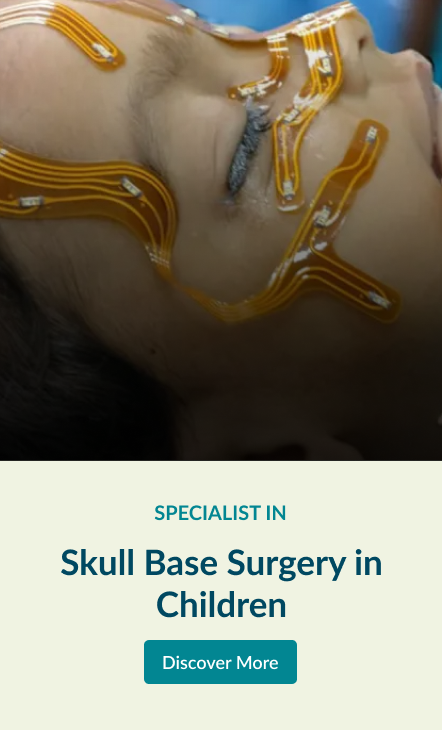
Skull Base Surgery in Children

Head and Neck Surgery
Head and neck cancers include cancers in the larynx (voice box), throat, lips, mouth, nose, and salivary glands. Because of their location, head and neck tumours may impair patients’ ability to eat, swallow and breathe. At Apollo Cancer Centre, Chennai, our multidisciplinary head and neck cancer experts works with patients to help them make informed decisions about their treatment options. Explore the links on this page to learn more about the different types of head and neck cancer, how they are treated. Learn how the head & neck surgeons, reconstructive surgeons and the speech and swallowing therapists work together to improve the patients’ ability to eat, swallow and breathe.
Read More
Open Skull Base Surgery
The skull base is the bony surface under the brain. There are number of structures lies below the skull base viz. nose & sinuses, eyes, ears etc. Major blood vessels taking blood to and from brain, nerves of the eye, ear and those controlling facial movement, swallowing & speech and spinal cord passes through number of natural foramina in the skull base. Tumours at the skull base are often close to critical areas of the brain and these nerves and blood vessels. This makes this area very complex.
(Apollo’s Comprehensive Skull Base Surgical team (established in 1995) consisting of ENT-Head & Neck Surgeons, Neurosurgeons and Plastic and reconstructive surgeons uses innovative surgical techniques to remove theses tumours in this complex area.
Explore the links on this page to learn more about the different aspects of open skull base surgery.
Read More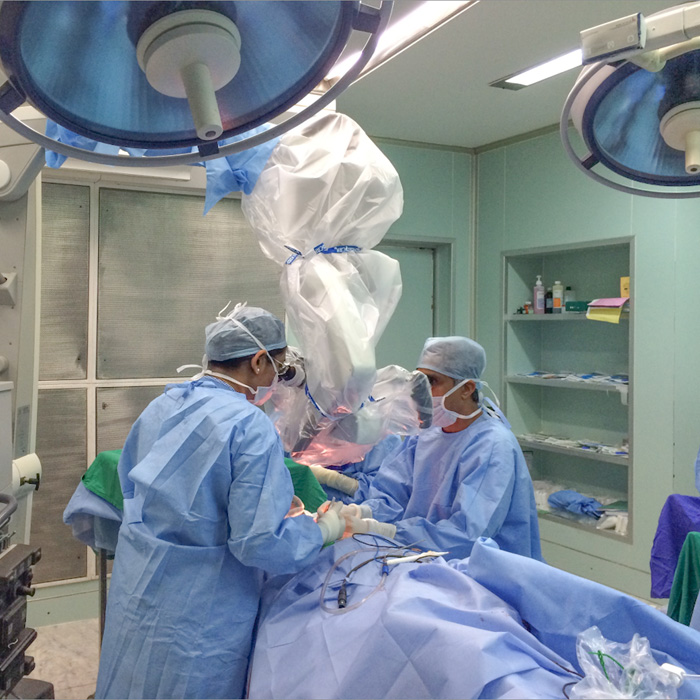
Lateral Skull Base Surgery
The “lateral skull base” is located at the side of the skull. This portion of the skull base includes structures called the temporal bone (most of the ear is within this bone), infratemporal fossa, clivus, and middle and posterior fossae.
Lateral skull base surgery can be used to treat conditions like meningiomas, acoustic neuromas, encephaloceles , cholesteatomas, schwannomas, glomus jugulare tumors and malignant tumours of the ear and temporal bone.
Explore the links on this page to learn more about the different aspects of lateral skull base surgery.
Read More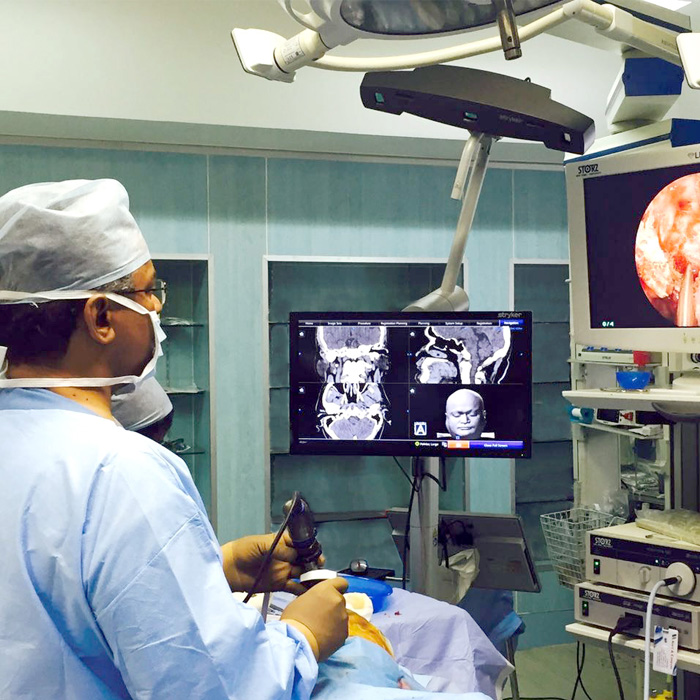
Endoscopic Pituitary Surgery
Pituitary gland is an endocrine gland which is located at the centre of the head under the brain close to sphenoid sinus. Pituitary tumours can cause hormone problems and vision loss. Endoscopic surgery is performed through the nose to remove tumours from the pituitary gland. In this minimally invasive surgery, the surgeon works through the nostrils with a tiny endoscope camera and light to remove tumours with special instruments. It gives better illumination and panoramic view of the pituitary gland and adjacent neuromuscular structures. This has resulted in better tumour clearance and lesser complications compared to the microscopic technique. Most of these tumours can be removed endoscopically without opening the skull (Craniotomy). Tumour removal often reverses vision problems and restores normal hormone balance.
Explore the links on this page to learn more about the different types of pituitary tumours, our technique and results.
Read More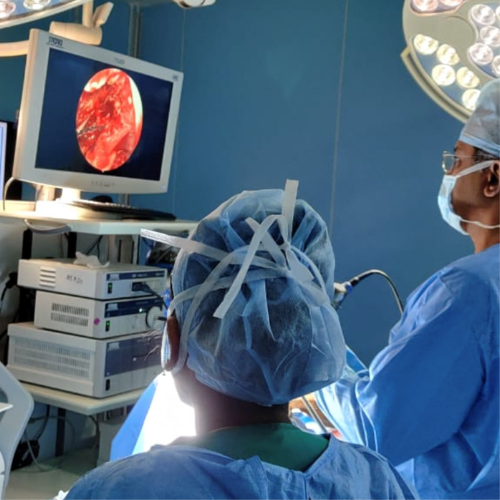
Endoscopic Skullbase Surgery
Endoscopic Skull Base Surgery is a form of minimally invasive surgery to correct a number of different conditions affecting the skull base (the region between the brain and roof & back of the nose)
Part of the skull base is related to nose, sinuses and nasopharynx. Tumours or tumour-like conditions in this region of skull base are treated endoscopically through the nose using different angled telescopes and special instruments with out opening the skull or making facial incisions.
A variety of conditions viz. CSF leak, meningoencephalocoele (herniation of brain inside the nose & sinuses), benign (inverted papilloma, nasopharyngeal angiofibroma) and malignant tumours of sinuses, meningiomas, clival chordoma, petrous apex lesions, nasopharyngeal carcinoma etc can be managed endoscopically through the nose
Explore the links on this page to learn more about the different aspects of endoscopic skull base surgery.
Read More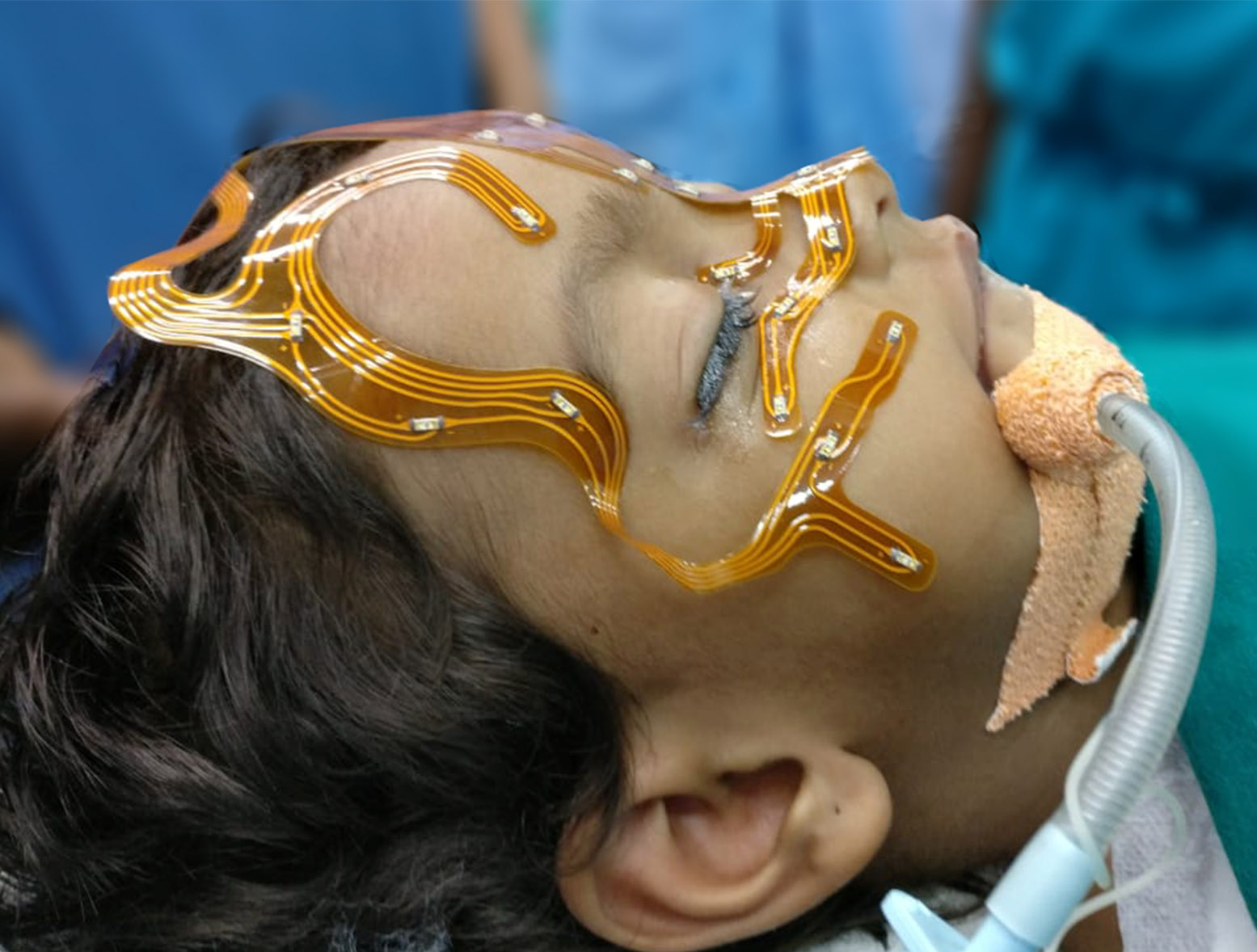
Pediateric Skull Base Surgery
Paediateric skull base surgery for tumours or tumour-like conditions is unique because of the small size of the growing skull and their inability withstand blood loss during surgery.
A variety of conditions viz. dermoid, teratoma (developmental tumour and contain keratin, cellular debris, hair, and sebum), herniation of brain, craniopharyngioma and malignant tumours (more likely sarcomas) occur in paediateric skull base.
Pediatric skull base surgery is a rapidly developing field involving multiple specialties. The collaboration between otolaryngology, neurosurgery, pediatric critical care and nursing has evolved to allow for the optimal management of pediatric patients.
Surgeons use innovative surgical techniques to remove theses tumours in this complex area of children with minimal effect in the normal growth of the skull. Our vast experience in adult open and endoscopic skull base surgery is safely implemented in treating these conditions in children. Explore the links on this page to learn more about the different aspects of open skull base surgery.
Read More-
Head & Neck Tumour Surgery
-
Open Skull Base Surgery
-
Lateral Skull Base Surgery
-
Endoscopic Pituitary Surgery
-
Endoscopic Skull Base Surgery
-
Skull Base Surgery in Children
Head and Neck Surgery
Head and neck cancers include cancers in the larynx (voice box), throat, lips, mouth, nose, and salivary glands. Because of their location, head and neck tumours may impair patients’ ability to eat, swallow and breathe. At Apollo Cancer Centre, Chennai, our multidisciplinary head and neck cancer experts works with patients to help them make informed decisions about their treatment options. Explore the links on this page to learn more about the different types of head and neck cancer, how they are treated. Learn how the head & neck surgeons, reconstructive surgeons and the speech and swallowing therapists work together to improve the patients’ ability to eat, swallow and breathe.
Read MoreOpen Skull Base Surgery
The skull base is the bony surface under the brain. There are number of structures lies below the skull base viz. nose & sinuses, eyes, ears etc. Major blood vessels taking blood to and from brain, nerves of the eye, ear and those controlling facial movement, swallowing & speech and spinal cord passes through number of natural foramina in the skull base. Tumours at the skull base are often close to critical areas of the brain and these nerves and blood vessels. This makes this area very complex.
(Apollo’s Comprehensive Skull Base Surgical team (established in 1995) consisting of ENT-Head & Neck Surgeons, Neurosurgeons and Plastic and reconstructive surgeons uses innovative surgical techniques to remove theses tumours in this complex area.
Explore the links on this page to learn more about the different aspects of open skull base surgery.
Read MoreLateral Skull Base Surgery
The “lateral skull base” is located at the side of the skull. This portion of the skull base includes structures called the temporal bone (most of the ear is within this bone), infratemporal fossa, clivus, and middle and posterior fossae.
Lateral skull base surgery can be used to treat conditions like meningiomas, acoustic neuromas, encephaloceles , cholesteatomas, schwannomas, glomus jugulare tumors and malignant tumours of the ear and temporal bone.
Explore the links on this page to learn more about the different aspects of lateral skull base surgery.
Read MoreEndoscopic Pituitary Surgery
Pituitary gland is an endocrine gland which is located at the centre of the head under the brain close to sphenoid sinus. Pituitary tumours can cause hormone problems and vision loss. Endoscopic surgery is performed through the nose to remove tumours from the pituitary gland. In this minimally invasive surgery, the surgeon works through the nostrils with a tiny endoscope camera and light to remove tumours with special instruments. It gives better illumination and panoramic view of the pituitary gland and adjacent neuromuscular structures. This has resulted in better tumour clearance and lesser complications compared to the microscopic technique. Most of these tumours can be removed endoscopically without opening the skull (Craniotomy). Tumour removal often reverses vision problems and restores normal hormone balance.
Explore the links on this page to learn more about the different types of pituitary tumours, our technique and results.
Read MoreEndoscopic Skullbase Surgery
Endoscopic Skull Base Surgery is a form of minimally invasive surgery to correct a number of different conditions affecting the skull base (the region between the brain and roof & back of the nose)
Part of the skull base is related to nose, sinuses and nasopharynx. Tumours or tumour-like conditions in this region of skull base are treated endoscopically through the nose using different angled telescopes and special instruments with out opening the skull or making facial incisions.
A variety of conditions viz. CSF leak, meningoencephalocoele (herniation of brain inside the nose & sinuses), benign (inverted papilloma, nasopharyngeal angiofibroma) and malignant tumours of sinuses, meningiomas, clival chordoma, petrous apex lesions, nasopharyngeal carcinoma etc can be managed endoscopically through the nose
Explore the links on this page to learn more about the different aspects of endoscopic skull base surgery.
Read MorePediateric Skull Base Surgery
Paediateric skull base surgery for tumours or tumour-like conditions is unique because of the small size of the growing skull and their inability withstand blood loss during surgery.
A variety of conditions viz. dermoid, teratoma (developmental tumour and contain keratin, cellular debris, hair, and sebum), herniation of brain, craniopharyngioma and malignant tumours (more likely sarcomas) occur in paediateric skull base.
Pediatric skull base surgery is a rapidly developing field involving multiple specialties. The collaboration between otolaryngology, neurosurgery, pediatric critical care and nursing has evolved to allow for the optimal management of pediatric patients.
Surgeons use innovative surgical techniques to remove theses tumours in this complex area of children with minimal effect in the normal growth of the skull. Our vast experience in adult open and endoscopic skull base surgery is safely implemented in treating these conditions in children. Explore the links on this page to learn more about the different aspects of open skull base surgery.
Read More


